Shen Lab Publications
Tamayo Jaramillo D., Hegde S., Jia X., Coffman, K., Vankayalaptai, H., Bearss, D. Jones, K.B., Stark, A.W., Shen PS.
Advanced Science. 2026 March 07
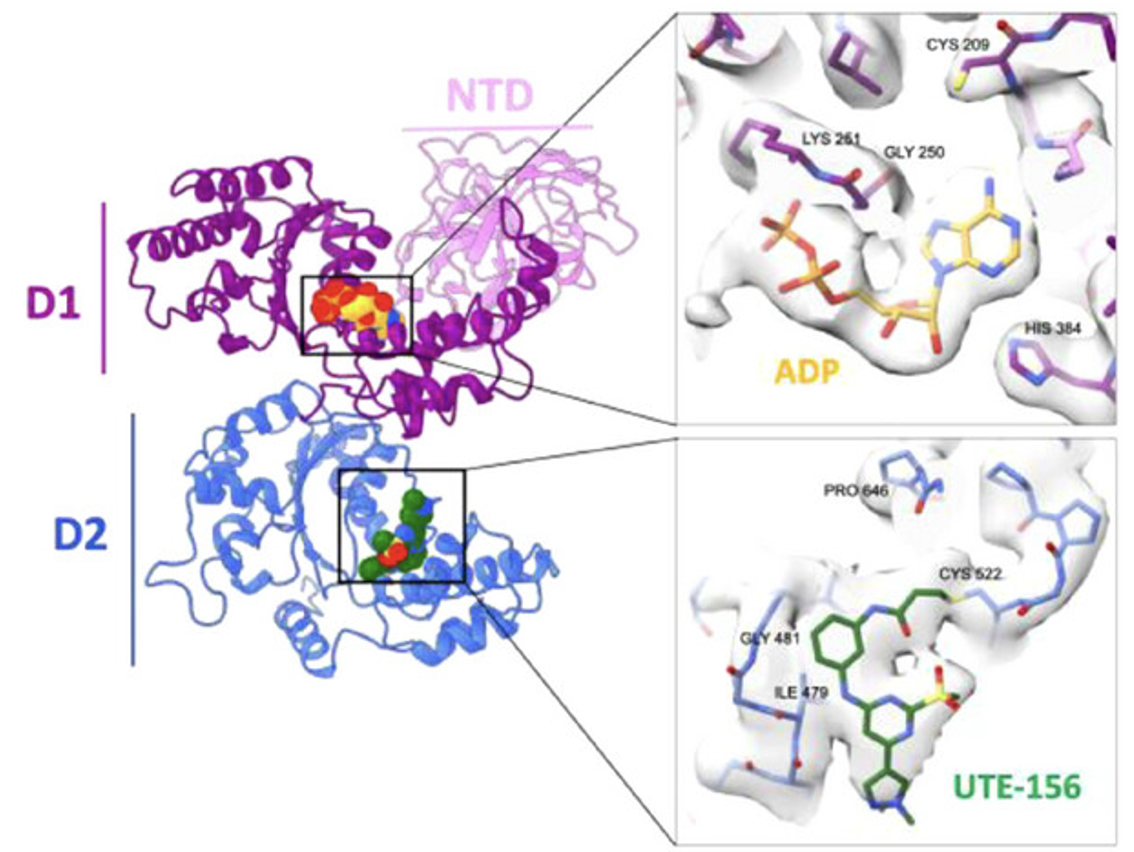
Development and Structural Characterization of UTE-156, a Covalent Inhibitor of the VCP/p97 AAA+ ATPase
The AAA+ ATPase valosin-containing protein (VCP/p97) is a central regulator of protein homeostasis that is well characterized for its role in extracting and remodeling ubiquitinated substrates. Dysregulation of VCP activity contributes to the pathogenesis of neurodegenerative diseases and cancer, making it an important therapeutic target. Here, we report the development and characterization of UTE-156, a novel covalent small-molecule inhibitor that modifies Cys522 within the D2 ATPase domain of VCP. UTE-156 potently inhibits VCP ATPase activity, while losing activity against a C522A mutant, supporting a covalent mechanism of action. High-resolution cryo-electron microscopy (cryo-EM) structures reveal that UTE-156 occupies the D2 nucleotide-binding site, sterically blocking ATP binding and inducing conformational remodeling of the pocket. Biochemical and cell-based assays demonstrate strong inhibitory potency but limited solubility and rapid metabolic turnover. These pharmacochemical limitations preclude immediate therapeutic use but underscore its value as a chemical probe. Together, these findings establish UTE-156 as a powerful tool for dissecting VCP function and provide a framework for future optimization of covalent modulators of protein homeostasis.
This works was done in collaboration with Kevin Jones and the University of Utah Therapeutics Accelerator Hub.
Li W, Scheel T, Shen PS
Nature Communications. 2025 July 01
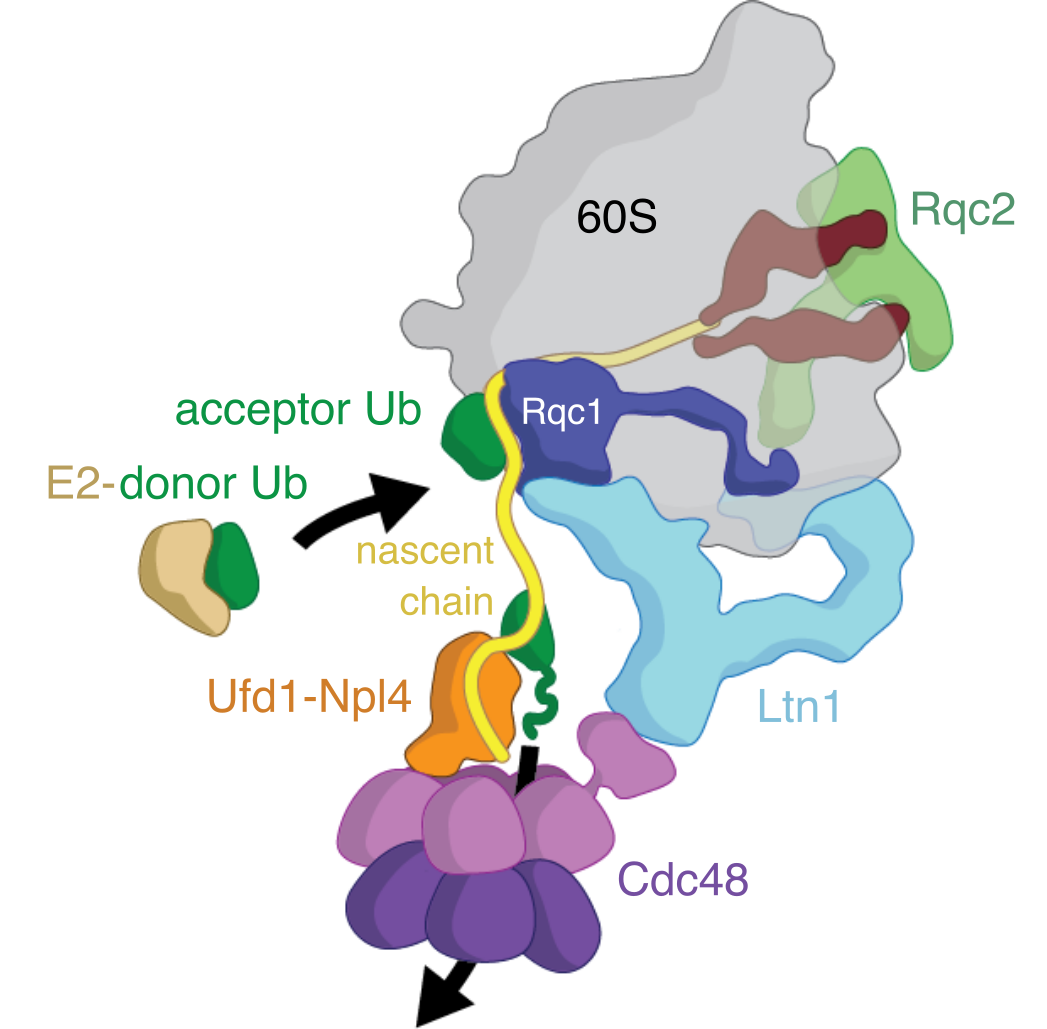
Mechanism of nascent chain removal by the ribosome-associated quality control complex
Errors during translation can cause ribosome stalling, leaving incomplete nascent chains attached to large ribosomal subunits. Cells rely on the Ribosome-associated Quality Control (RQC) complex to recognize, process, and remove these aberrant proteins to maintain proteostasis. Despite its importance, the mechanisms by which the RQC orchestrates nascent chain processing and extraction have remained unclear. Here, we present a cryo-EM structure of the RQC complex from budding yeast, revealing how its core components function in nascent chain removal. We show that the Cdc48 ATPase and its Ufd1-Npl4 adaptor are recruited by the Ltn1 E3 ubiquitin ligase to extract ubiquitylated peptides from the 60S ribosome. Additionally, we find that Rqc1 bridges the 60S subunit with ubiquitin and Ltn1, facilitating formation of K48-linked polyubiquitin chains. These findings provide a structural and mechanistic framework for understanding how the RQC complex collaborates to clear stalled translation products, advancing insight into cellular protein quality control.
Aderounmu AM, Maus-Conn J, Consalvo CD, Shen PS#, Bass BL#
PNAS. 2025 April 28
# co-corresponding authors
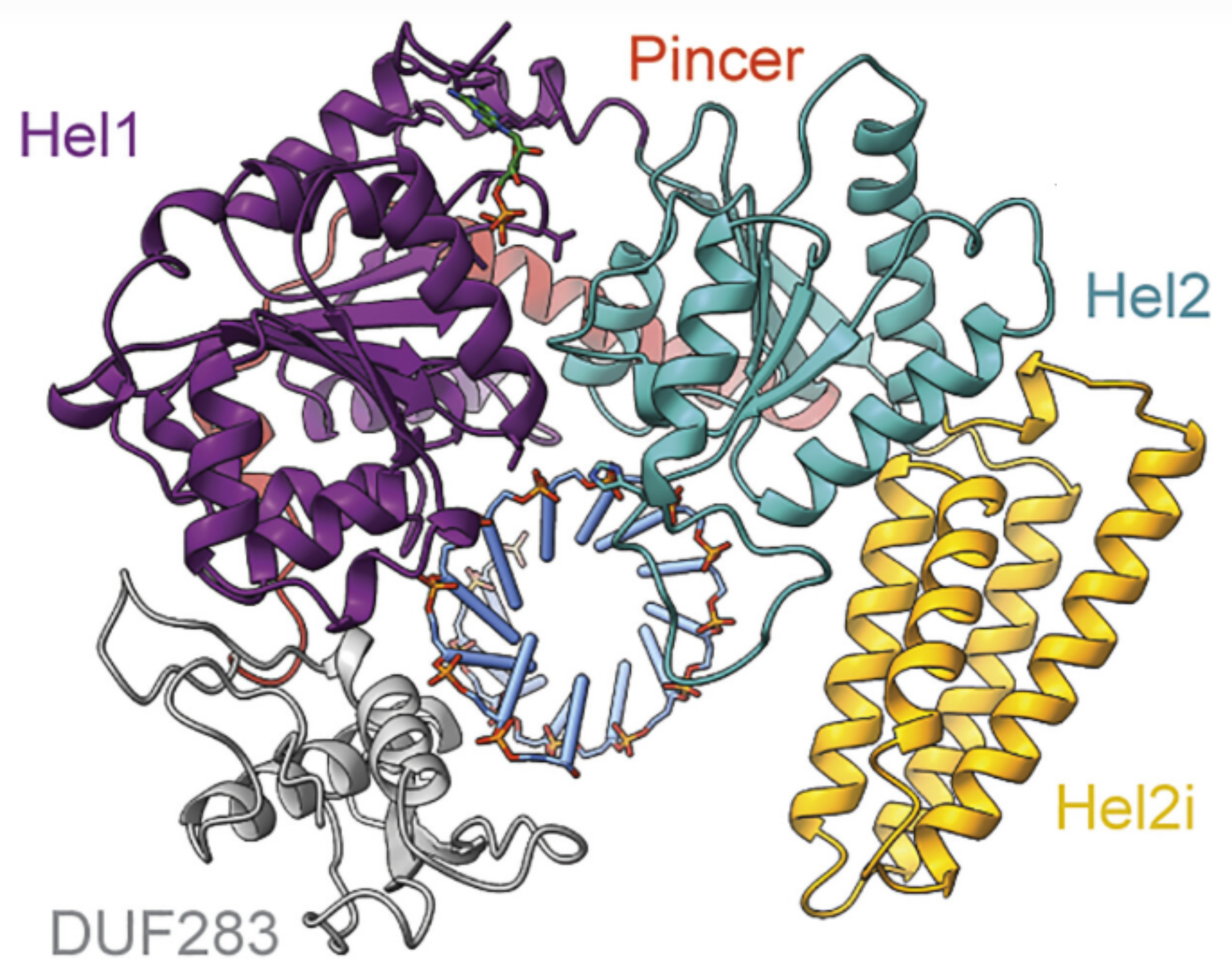
Biochemical and structural basis of Dicer helicase function unveiled by resurrecting ancient proteins
Among animals, the contribution of Dicer’s helicase to recognition and elimination of viral double-stranded RNA (dsRNA) varies from phylum to phylum. Vertebrate Dicers show no helicase activity, while an arthropod ortholog uses helicase translocation to efficiently move dsRNA into Dicer’s cleavage site. The biochemical and structural basis of Dicer’s helicase function, and the evolutionary events that contribute to divergence in function, have remained unknown. This study shows how ancient Dicer helicase tightly binds dsRNA and couples adenosine triphosphate (ATP) hydrolysis to movement along dsRNA. The data reveal how components of this intricate system declined along different clades of animal evolution.
This work was done in collaboration with Brenda Bass.

Cooney I*, Schubert HL*, Cedeno K, Fisher ON, Carson R, Price JC, Hill CP#, Shen PS#
Nature Communications. 2024 August 29
*co-first authors
# co-corresponding authors
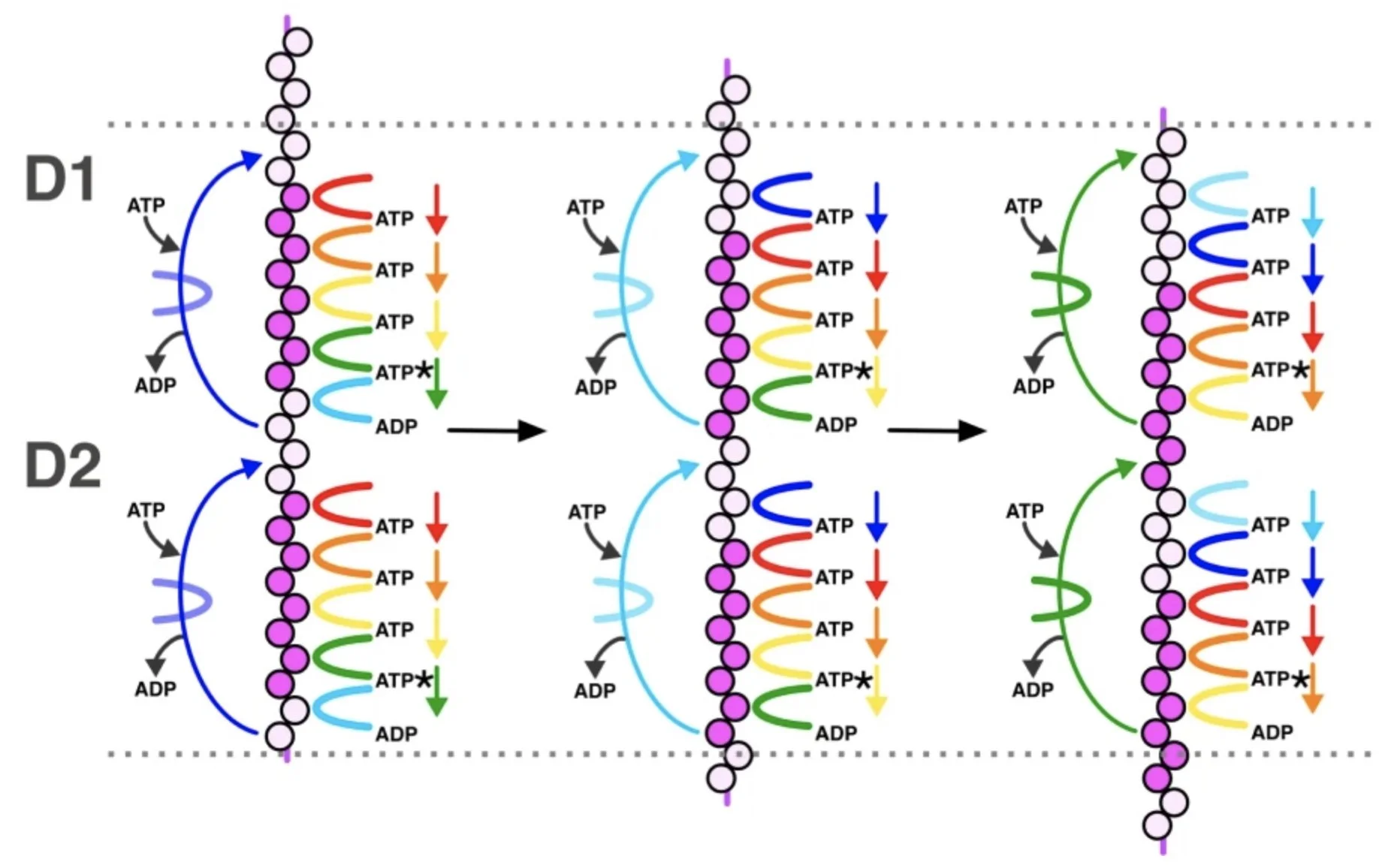
Visualization of the Cdc48 AAA+ ATPase protein unfolding pathway
The Cdc48 AAA+ ATPase is an abundant and essential enzyme that unfolds substrates in multiple protein quality control pathways. The enzyme includes two conserved AAA+ ATPase motor domains, D1 and D2, that assemble as hexameric rings with D1 stacked above D2. Here, we report an ensemble of native structures of Cdc48 affinity purified from budding yeast lysate in complex with the adaptor Shp1 in the act of unfolding substrate. Our analysis reveals a continuum of structural snapshots that spans the entire translocation cycle. These data uncover elements of Shp1-Cdc48 interactions and support a ‘hand-over-hand’ mechanism in which the sequential movement of individual subunits is closely coordinated. D1 hydrolyzes ATP and disengages from substrate prior to D2, while D2 rebinds ATP and re-engages with substrate prior to D1, thereby explaining the dominant role played by the D2 motor in substrate translocation/unfolding.
This work was done in collaboration with Chris Hill.
Consalvo CD, Aderounmu AM, Donelick HM, Aruscavage PJ, Eckert DM, Shen PS*, Bass BL*
eLife. 2024 April 3
*co-corresponding authors
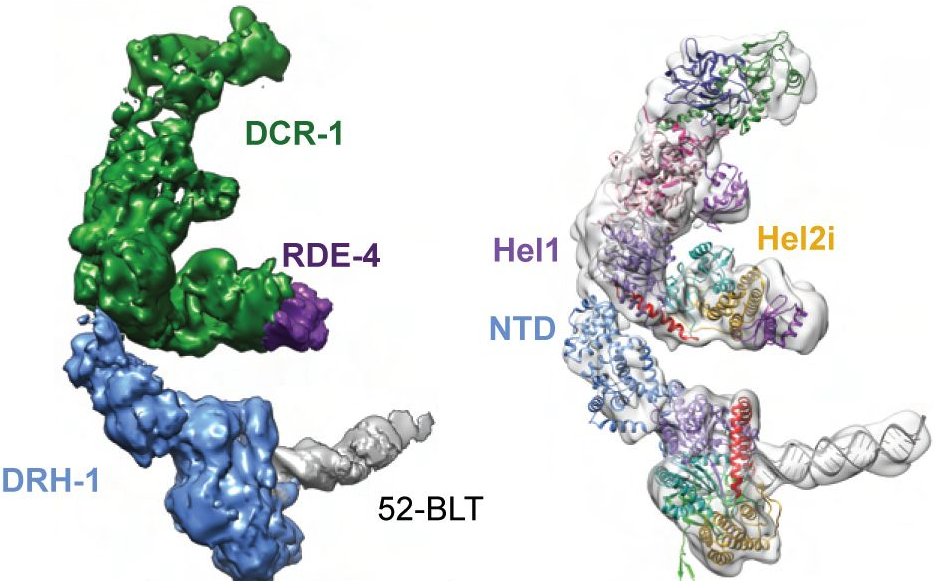
C. elegans Dicer acts with the RIG-I-like helicase DRH-1 and RDE-4 to cleave dsRNA
Invertebrates use the endoribonuclease Dicer to cleave viral dsRNA during antiviral defense, while vertebrates use RIG-I-like Receptors (RLRs), which bind viral dsRNA to trigger an interferon response. While some invertebrate Dicers act alone during antiviral defense, C. elegans Dicer acts in a complex with a dsRNA binding protein called RDE-4, and an RLR ortholog called DRH-1. We used biochemical and structural techniques to provide mechanistic insight into how these proteins function together. We found RDE-4 is important for ATP-independent and ATP-dependent cleavage reactions, while helicase domains of both DCR-1 and DRH-1 contribute to ATP-dependent cleavage. DRH-1 plays the dominant role in ATP hydrolysis, and like mammalian RLRs, has an N-terminal domain that functions in autoinhibition. A cryo-EM structure indicates DRH-1 interacts with DCR-1’s helicase domain, suggesting this interaction relieves autoinhibition. Our study unravels the mechanistic basis of the collaboration between two helicases from typically distinct innate immune defense pathways.
This work was done in collaboration with Brenda Bass.

Wang S*, Sass MI*, Kwon Y, Ludlam WG, Smith TM, Carter EJ, Gladden NE, Riggi M, Iwasa JH, Willardson BM#, Shen PS#
Molecular Cell. 2023 Nov 2
*co-first authors
#co-corresponding authors
Visualizing the chaperone-mediated folding trajectory of the G protein beta5 beta-propeller
The Chaperonin Containing Tailless polypeptide 1 (CCT) complex is an essential protein folding machine with a diverse clientele of substrates, including many proteins with β-propeller domains. Here, we determine the structures of human CCT in complex with its accessory co-chaperone, phosducin-like protein 1 (PhLP1), in the process of folding Gβ5, a component of Regulator of G protein Signaling (RGS) complexes. Cryoelectron microscopy (cryo-EM) and image processing reveal an ensemble of distinct snapshots that represent the folding trajectory of Gβ5 from an unfolded molten globule to a fully folded β-propeller. These structures reveal the mechanism by which CCT directs Gβ5 folding through initiating specific intermolecular contacts that facilitate the sequential folding of individual β sheets until the propeller closes into its native structure. This work directly visualizes chaperone-mediated protein folding and establishes that CCT orchestrates folding by stabilizing intermediates through interactions with surface residues that permit the hydrophobic core to coalesce into its folded state.
This work was done in collaboration with Barry Willardson.

Cooney I, Mack DC, Ferrell AJ, Stewart MG, Wang S, Donelick HM, Tamayo-Jaramillo D, Greer DL, Zhu D, Li W, Shen PS.
Bio-protocol. 2023 Jan 20
Lysate-to-grid: Rapid Isolation of Native Complexes from Budding Yeast for Cryo-EM Imaging
Single-particle electron cryo-microscopy (cryo-EM) is an effective tool to determine high-resolution structures of macromolecular complexes. Its lower requirements for sample concentration and purity make it an accessible method to determine structures of low-abundant protein complexes, such as those isolated from native sources. While there are many approaches to protein purification for cryo-EM, attaining suitable particle quality and abundance is generally the major bottleneck to the typical single-particle project workflow. Here, we present a protocol using budding yeast ( S. cerevisiae ), in which a tractable immunoprecipitation tag (3xFLAG) is appended at the endogenous locus of a gene of interest (GOI). The modified gene is expressed under its endogenous promoter, and cells are grown and harvested using standard procedures. Our protocol describes the steps in which the tagged proteins and their associated complexes are isolated within three hours of thawing cell lysates, after which the recovered proteins are used directly for cryo-EM specimen preparation. The prioritization of speed maximizes the ability to recover intact, scarce complexes. The protocol is generalizable to soluble yeast proteins that tolerate C-terminal epitope tags.
Xu Y*, Han H*, Cooney I, Guo Y, Moran NG, Zuniga NR, Price JC, Hill CP#, Shen PS#
Nature Communications. 12 May 2022
*Co-first authors
#Co-corresponding authors
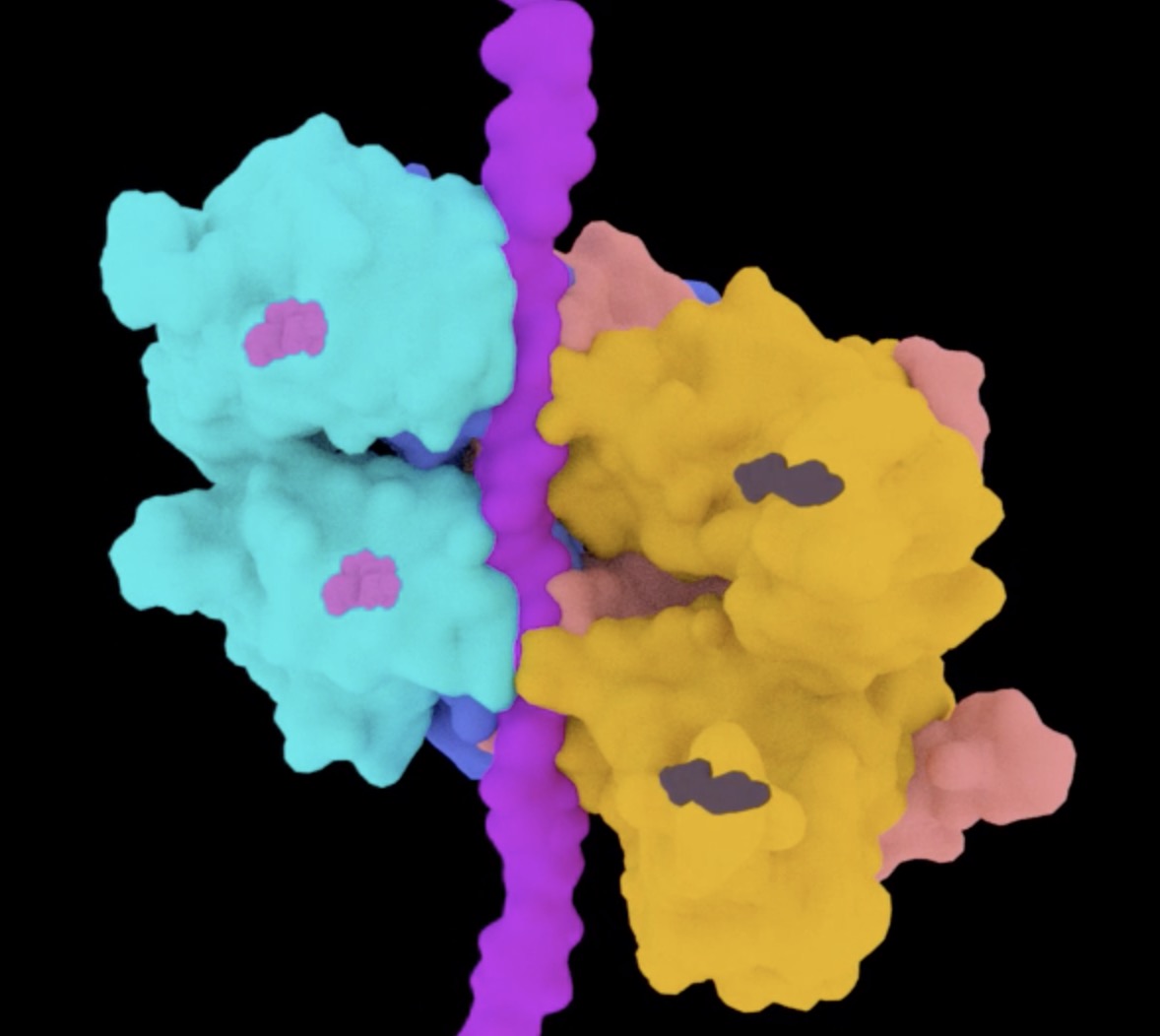
Active conformation of the p97-p47 unfoldase complex
The p97 AAA+ATPase is an essential and abundant regulator of protein homeostasis that plays a central role in unfolding ubiquitylated substrates. Here we report two cryo-EM structures of human p97 in complex with its p47 adaptor. One of the conformations is six-fold symmetric, corresponds to previously reported structures of p97, and lacks bound substrate. The other structure adopts a helical conformation, displays substrate running in an extended conformation through the pore of the p97 hexamer, and resembles structures reported for other AAA unfoldases. These findings support the model that p97 utilizes a “hand-over-hand” mechanism in which two residues of the substrate are translocated for hydrolysis of two ATPs, one in each of the two p97 AAA ATPase rings. Proteomics analysis supports the model that one p97 complex can bind multiple substrate adaptors or binding partners, and can process substrates with multiple types of ubiquitin modification.
This work was done in collaboration with Chris Hill.
Rollins MG, Shasmal M, Meade N, Astar H, Shen PS*, Walsh D* (*co-corresponding authors)
Cell Reports. 7 Sep 2021
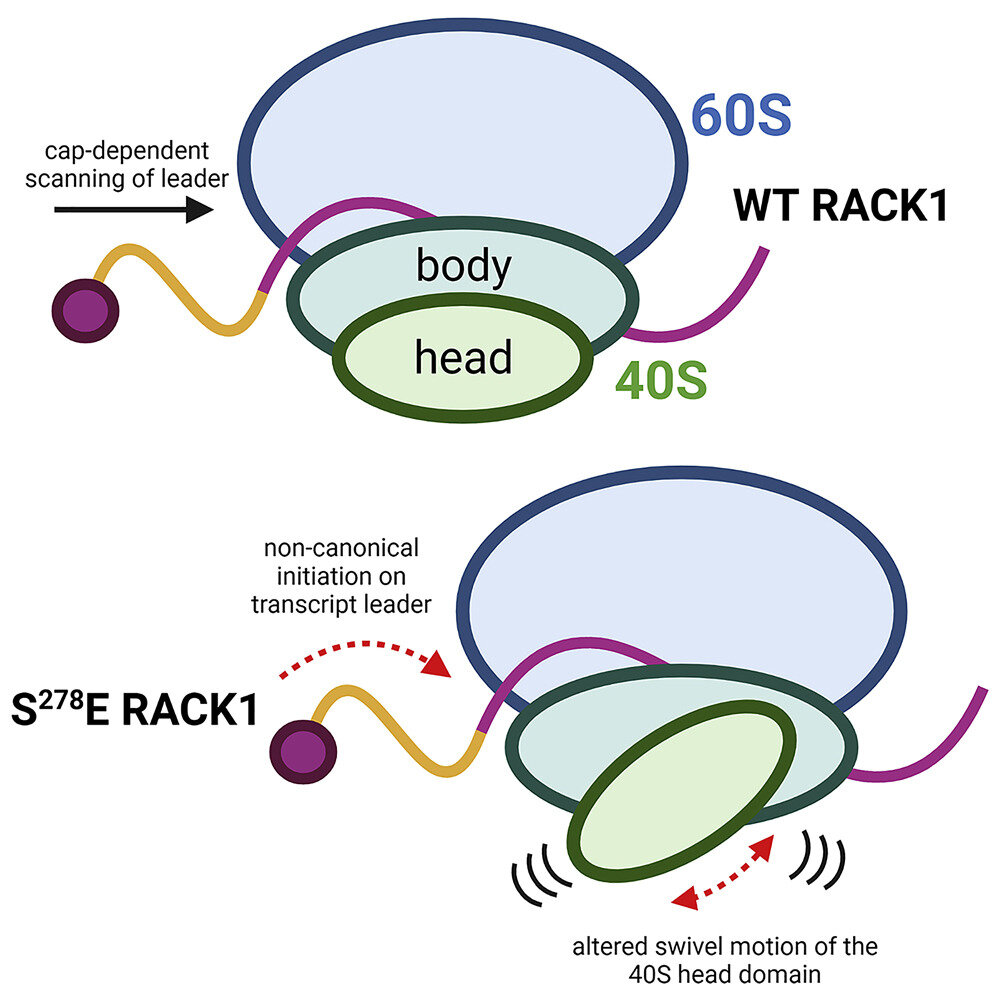
Negative charge in the RACK1 loop broadens the translational capacity of the human ribosome
Although the roles of initiation factors, RNA binding proteins, and RNA elements in regulating translation are well defined, how the ribosome functionally diversifies remains poorly understood. In their human hosts, poxviruses phosphorylate serine 278 (S278) at the tip of a loop domain in the small subunit ribosomal protein RACK1, thereby mimicking negatively charged residues in the RACK1 loops of dicot plants and protists to stimulate translation of transcripts with 5' poly(A) leaders. However, how a negatively charged RACK1 loop affects ribosome structure and its broader translational output is not known. Here, we show that although ribotoxin-induced stress signaling and stalling on poly(A) sequences are unaffected, negative charge in the RACK1 loop alters the swivel motion of the 40S head domain in a manner similar to several internal ribosome entry sites (IRESs), confers resistance to various protein synthesis inhibitors, and broadly supports noncanonical modes of translation.
This work was done in collaboration with Derek Walsh.
Cooney I*, Han H*, Stewart MG, Carson RH, Hansen DT, Iwasa JH, Price JC, Hill CP†, Shen PS† (*equal contributing authors; †co-corresponding authors)
Science. 27 Jun 2019 (First Release)
Structure of the Cdc48 segregase in the act of unfolding an authentic substrate
The cellular machine Cdc48 functions in multiple biological pathways by segregating its protein substrates from a variety of stable environments such as organelles or multi-subunit complexes. Despite extensive studies, the mechanism of Cdc48 has remained obscure, and its reported structures are inconsistent with models of substrate translocation proposed for other AAA+ ATPases. Here, we report a 3.7 Å resolution structure of Cdc48 in complex with an adaptor protein and a native substrate. Cdc48 engages substrate by adopting a helical configuration of substrate-binding residues that extends through the central pore of both of the ATPase rings. These findings indicate a unified hand-over-hand mechanism of protein translocation by Cdc48 and other AAA+ ATPases.
This work was done in collaboration with Chris Hill.

Han H, Fulcher JM, Dandey VP, Iwasa JH, Sundquist WI, Kay MS, Shen PS*, Hill CP* (*co-corresponding authors)
Elife. 2019 Jun 11;8. pii: e44071
Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases
Many AAA+ ATPases form hexamers that unfold protein substrates by translocating them through their central pore. Multiple structures have shown how a helical assembly of subunits binds a single strand of substrate, and indicate that translocation results from the ATP-driven movement of subunits from one end of the helical assembly to the other end. To understand how more complex substrates are bound and translocated, we demonstrated that linear and cyclic versions of peptides bind to the S. cerevisiae AAA+ ATPase Vps4 with similar affinities, and determined cryo-EM structures of cyclic peptide complexes. The peptides bind in a hairpin conformation, with one primary strand equivalent to the single chain peptide ligands, while the second strand returns through the translocation pore without making intimate contacts with Vps4. These observations indicate a general mechanism by which AAA+ ATPases may translocate a variety of substrates that include extended chains, hairpins, and crosslinked polypeptide chains.
This work was done in collaboration with Chris Hill.

Sinha NK, Iwasa JH, Shen PS*, Bass BL* (*co-corresponding authors)
Science. 2018 Jan 19;359(6373):329-334.
Dicer uses distinct modules for recognizing dsRNA termini.
Invertebrates rely on Dicer to cleave viral double-stranded RNA (dsRNA), and Drosophila Dicer-2 distinguishes dsRNA substrates by their termini. Blunt termini promote processive cleavage, while 3' overhanging termini are cleaved distributively. To understand this discrimination, we used cryo-electron microscopy to solve structures of Drosophila Dicer-2 alone and in complex with blunt dsRNA. Whereas the Platform-PAZ domains have been considered the only Dicer domains that bind dsRNA termini, unexpectedly, we found that the helicase domain is required for binding blunt, but not 3' overhanging, termini. We further showed that blunt dsRNA is locally unwound and threaded through the helicase domain in an adenosine triphosphate-dependent manner. Our studies reveal a previously unrecognized mechanism for optimizing antiviral defense and set the stage for the discovery of helicase-dependent functions in other Dicers.
This work was done in collaboration with Brenda Bass.

Han H, Monroe N, Sundquist WI*, Shen PS*, Hill CP* (co-corresponding authors)
Elife. 2017 Nov 22;6. pii: e31324.
The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets
The hexameric AAA ATPase Vps4 drives membrane fission by remodeling and disassembling ESCRT-III filaments. Building upon our earlier 4.3 Å resolution cryo-EM structure (Monroe et al., 2017), we now report a 3.2 Å structure of Vps4 bound to an ESCRT-III peptide substrate. The new structure reveals that the peptide approximates a β-strand conformation whose helical symmetry matches that of the five Vps4 subunits it contacts directly. Adjacent Vps4 subunits make equivalent interactions with successive substrate dipeptides through two distinct classes of side chain binding pockets formed primarily by Vps4 pore loop 1. These pockets accommodate a wide range of residues, while main chain hydrogen bonds may help dictate substrate-binding orientation. The structure supports a ‘conveyor belt’ model of translocation in which ATP binding allows a Vps4 subunit to join the growing end of the helix and engage the substrate, while hydrolysis and release promotes helix disassembly and substrate release at the lagging end.
This work was done in collaboration with Chris Hill and Wes Sundquist.

Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, Brandman O, Frost A.
Science. 2015 Jan 2;347(6217):75-8.
Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains
In Eukarya, stalled translation induces 40S dissociation and recruitment of the ribosome quality control complex (RQC) to the 60S subunit, which mediates nascent chain degradation. Here we report cryo–electron microscopy structures revealing that the RQC components Rqc2p and Ltn1p bind to the 60S subunit at sites exposed after 40S dissociation, placing the Ltn1p RING domain near the exit channel and Rqc2p over the P-site tRNA. We further demonstrate that Rqc2p recruits alanine- and threonine-charged tRNA to the A site and directs the elongation of nascent chains independently of mRNA or 40S subunits. Our work uncovers an unexpected mechanism of protein synthesis, in which a protein—not an mRNA—determines tRNA recruitment and the tagging of nascent chains with carboxy-terminal Ala and Thr extensions (“CAT tails”).
This work was done by a collaboration among the Frost, Brandman, and Weissman labs.